Ischémie myocardique et infarctus du myocarde : modifications cellulaires, ECG et symptômes
Ischémie myocardique : modifications cellulaires, signes ECG et développement de l’infarctus
- La cascade ischémique : chronologie des événements
- L’ischémie myocardique en pratique clinique : la maladie coronarienne
- Physiopathologie des modifications électriques : Les courants de lésion
- Le temps est du muscle : 30 minutes entre l’ischémie myocardique et l’infarctus
- Vulnérabilité du sous-endocarde
- Phénomènes post-ischémiques : Sidération et Hibernation
Comme toutes les autres cellules du corps humain, les myocytes cardiaques utilisent l’ATP (adénosine triphosphate) comme principale source d’énergie. À l’état basal, le cœur est un organe omnivore mais privilégie l’oxydation des acides gras libres (60 à 90 % de l’énergie) par rapport au glucose et aux lactates. L’ATP est produit par le métabolisme mitochondrial oxydatif, nécessitant un apport constant en oxygène. L’ATP alimente toutes les fonctions cellulaires, notamment le maintien des gradients ioniques transmembranaires (via la pompe Na+/K+ ATPase) et le couplage excitation-contraction (cycle du calcium et glissement des myofilaments d’actine et de myosine).
L’augmentation de la charge de travail cardiaque par l’augmentation de la fréquence cardiaque (chronotropisme positif) et de la contractilité (inotropisme positif) entraîne une augmentation linéaire de la consommation myocardique en oxygène (MVO2). Les cellules cardiaques saines régulent finement l’offre et la demande d’ATP : une augmentation de la demande entraîne une vasodilatation coronaire métabolique (hyperhémie fonctionnelle) pour accroître l’apport en substrats et en oxygène. Il est crucial de noter que l’extraction d’oxygène par le myocarde est quasi-maximale au repos (environ 70-80 %), contrairement aux muscles squelettiques. Par conséquent, toute augmentation de la demande doit être satisfaite par une augmentation proportionnelle du flux sanguin coronaire.
L’ischémie myocardique survient lorsqu’il existe un déséquilibre entre l’apport (perfusion) et la demande en oxygène. L’hypoxie cellulaire entraîne une chute rapide de la production d’ATP mitochondrial et une perturbation de l’homéostasie ionique. En cas d’ischémie, les myocytes cardiaques tentent de préserver leur viabilité en basculant vers la glycolyse anaérobie. Cela est possible grâce aux réserves de glycogène intracellulaire. Cependant, ce processus est énergétiquement inefficace, produisant nettement moins d’ATP et générant des protons ($H^+$) et du lactate. L’acidose intracellulaire résultante inhibe les protéines contractiles et les canaux ioniques.
Afin de réduire drastiquement la consommation d’oxygène et préserver l’ATP pour les fonctions vitales (maintien de l’intégrité membranaire), les myocytes cessent de se contracter : c’est le couplage perfusion-contraction. Ces mesures adaptatives – glycolyse anaérobie et arrêt de la contraction – définissent une fenêtre de survie de 20 à 30 minutes d’ischémie sévère. Au-delà, l’épuisement total de l’ATP entraîne l’arrêt des pompes ioniques, une surcharge calcique massive, un œdème cellulaire et la rupture des membranes, marquant le début de la nécrose de coagulation irréversible (mort cellulaire).

La cascade ischémique : chronologie des événements
Il est fondamental pour le clinicien de comprendre que l’ischémie myocardique déclenche une séquence d’événements prévisible, connue sous le nom de « cascade ischémique ». L’expression clinique (douleur) et électrique (ECG) sont des phénomènes tardifs dans cette séquence :
- Perturbations métaboliques : Passage au métabolisme anaérobie, acidose, accumulation de lactate.
- Dysfonction diastolique : La relaxation est un processus actif consommant de l’ATP (recapture du calcium par le réticulum sarcoplasmique). L’ischémie provoque précocement une rigidité du ventricule gauche (baisse de la compliance) et une élévation des pressions de remplissage.
- Dysfonction systolique : Hypokinésie, puis akinésie ou dyskinésie de la zone affectée.
- Modifications électrocardiographiques : Altérations de la repolarisation (ondes T, segment ST).
- Symptômes cliniques : Angor (douleur thoracique), dyspnée.
Cette séquence explique pourquoi l’imagerie (échocardiographie de stress) peut détecter une ischémie plus précocement ou avec une plus grande sensibilité que l’ECG d’effort seul.
L’ischémie myocardique en pratique clinique : la maladie coronarienne
En pratique clinique, l’ischémie myocardique se manifeste principalement par la maladie coronarienne athéroscléreuse, bien que d’autres causes (spasme, dissection, embolie) existent. On distingue classiquement deux physiopathologies principales :
Dans le syndrome coronarien chronique (angine stable), des plaques d’athérome fibro-lipidiques réduisent la lumière vasculaire. Au repos, le flux peut être suffisant. Cependant, lors d’un effort physique ou d’un stress émotionnel, l’incapacité à augmenter le flux à travers la sténose crée une ischémie d’effort (inadéquation offre/demande). Plus la sténose est serrée, plus le seuil ischémique est bas. L’épreuve d’effort vise à reproduire ce déséquilibre pour démasquer les modifications ECG (sous-décalage ST) et les symptômes.
Dans les syndromes coronariens aigus (SCA), le mécanisme est différent. Il s’agit généralement d’une rupture ou d’une érosion de plaque d’athérome entraînant une thrombose intra-luminale brutale. Cette occlusion partielle ou totale réduit le flux sanguin de manière critique, indépendamment de la demande myocardique. Les symptômes et les signes ECG apparaissent alors au repos. Si l’occlusion est complète et persistante, elle conduit à un infarctus avec élévation du segment ST (STEMI). Si elle est partielle ou intermittente, elle conduit à un SCA sans élévation du segment ST (NSTEMI) ou à un angor instable.
Tableau 1 : Réaction myocardique et ECG dans différents contextes d’ischémie ou d’augmentation de la charge de travail
| État de l’artère coronaire | SITUATION | EFFET PHYSIOPATHOLOGIQUE | Réaction ECG |
| Artère coronaire normale (pas d’athérosclérose) | Au repos | L’apport d’oxygène est adéquat et l’ischémie ne peut se produire. | Aucune modification de l’ECG au repos. |
| Augmentation de la charge de travail du myocarde (exercice) | L’apport d’oxygène augmente parallèlement à la consommation (vasodilatation métabolique). L’offre et la demande sont équilibrées. | Aucun changement pathologique.
Note : Une dépression jonctionnelle du segment ST avec pente ascendante rapide est physiologique à l’effort intense (tachycardie), sans signification ischémique. |
|
| Maladie coronarienne stable (sténose significative > 70 %) | Au repos | Le flux de repos est maintenu grâce à la dilatation des artérioles distales (réserve coronaire entamée mais suffisante au repos). Pas de symptômes. | Pas de modification de l’ECG au repos (sauf séquelles anciennes). |
| Augmentation de la charge de travail du myocarde (exercice) | La réserve coronaire est épuisée. La demande excède l’apport limité par la sténose fixe. L’ischémie se développe initialement dans le sous-endocarde (zone la plus vulnérable). C’est une ischémie dite « de demande ». | L’épreuve d’effort objective l’ischémie sous-endocardique par des sous-décalages du segment ST (horizontal ou descendant) et/ou des inversions de l’onde T. Ces signes régressent au repos. | |
| Maladie coronarienne stable sévère (sténose critique > 90 %) | Au repos | La sténose est telle que le flux de repos est à peine suffisant ou déjà compromis. Le myocarde peut être en état d’hibernation pour survivre. | L’ECG au repos peut montrer des anomalies de repolarisation persistantes (sous-décalage ST, ondes T négatives). |
| Rupture de plaque (Syndrome Coronarien Aigu) | À tout moment (souvent repos) | Ischémie transmurale (Occlusion totale) : Lésion de toute l’épaisseur du myocarde. Ischémie sous-endocardique (Occlusion subtotale) : Lésion limitée aux couches profondes. |
Occlusion totale : Élévation du segment ST (courant de lésion vers l’épicarde). Occlusion partielle : Sous-décalage du segment ST, ondes T négatives profondes. |
| Vasospasme coronaire (Angor de Prinzmetal) | Vasospasme transitoire | Une vasoconstriction sévère et focale de l’épicarde provoque une ischémie transmurale brutale mais réversible, souvent sans athérosclérose obstructive. | L’ECG enregistre des élévations transitoires du segment ST (image de STEMI réversible) qui disparaissent avec la résolution du spasme (ou trinitrine). |
Physiopathologie des modifications électriques : Les courants de lésion
Pourquoi l’ischémie modifie-t-elle le segment ST ? Les modifications de l’ECG résultent de gradients de voltage entre le tissu sain et le tissu ischémique. L’ischémie entraîne une ouverture des canaux potassiques ATP-dépendants et une perte du potentiel de repos membranaire (la cellule ischémique est partiellement dépolarisée au repos).
Cela génère des « courants de lésion ». En cas d’ischémie transmurale (occlusion totale), le vecteur de lésion se dirige de l’endocarde vers l’épicarde, entraînant une élévation du segment ST dans les dérivations faisant face à la zone ischémique. En cas d’ischémie sous-endocardique, le vecteur global se dirige vers la cavité ventriculaire (loin des électrodes de surface), ce qui se traduit par un sous-décalage du segment ST.
Le temps est du muscle : 30 minutes entre l’ischémie myocardique et l’infarctus
La durée de l’ischémie est le déterminant principal de la taille de l’infarctus final. Le myocarde alimenté par l’artère occluse (la « zone à risque ») devient immédiatement ischémique et cesse de se contracter (akinésie). Comme indiqué plus haut, les cellules maintiennent leur intégrité membranaire pendant une « période d’or » de 20 à 30 minutes grâce au métabolisme anaérobie.
Si le flux coronaire est rétabli (reperfusion spontanée ou thérapeutique par angioplastie/thrombolyse) au cours de cette période, la nécrose peut être évitée et le myocarde peut récupérer, bien qu’il puisse passer par une phase de dysfonction temporaire (sidération). Si l’occlusion persiste au-delà de 30 minutes, le front de nécrose s’installe. Ce processus suit le « phénomène de front d’onde » décrit par Reimer et Jennings : la mort cellulaire commence dans la zone la plus vulnérable, le sous-endocarde, et progresse vers l’épicarde en fonction de la durée de l’occlusion. Reportez-vous à la figure 2.
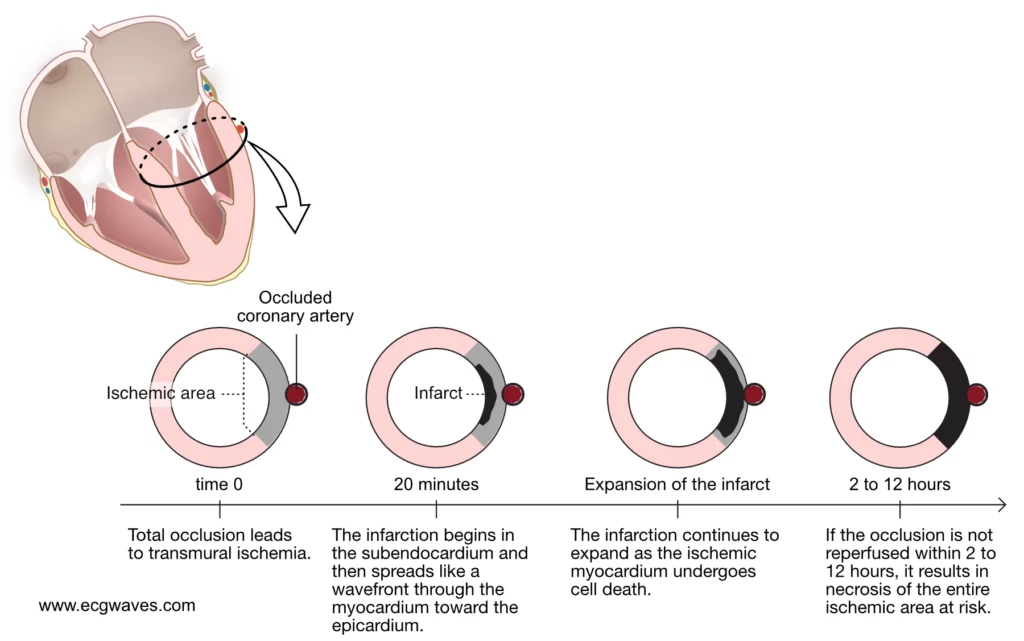
Le concept de « fenêtre thérapeutique » découle de cette progression. Bien que l’on considère classiquement que l’infarctus est constitué en 4 à 6 heures, ce délai est très variable (de 2 à 12 heures) selon les patients. Les facteurs protecteurs qui ralentissent la progression de la nécrose incluent :
- La circulation collatérale : Des anastomoses préexistantes peuvent perfuser la zone ischémique à contre-courant, limitant l’extension de l’infarctus.
- Le préconditionnement ischémique : Des épisodes brefs d’angine précédant l’infarctus peuvent rendre les myocytes plus résistants à l’hypoxie prolongée.
- La demande métabolique : Une fréquence cardiaque basse et une pression artérielle contrôlée réduisent la consommation d’oxygène et ralentissent la nécrose.
Les occlusions totales responsables de STEMI nécessitent une reperfusion urgente. Même si une reperfusion tardive ne sauve pas tout le myocarde, elle permet de limiter l’expansion de l’infarctus, de réduire le risque de rupture cardiaque et d’améliorer le remodelage ventriculaire à long terme (moins de dilatation, meilleure cicatrisation). Cependant, la reperfusion elle-même peut causer des dommages, appelés lésions de reperfusion (génération de radicaux libres, surcharge calcique), bien que le bénéfice net de la réouverture de l’artère reste incontestable.
Vulnérabilité du sous-endocarde
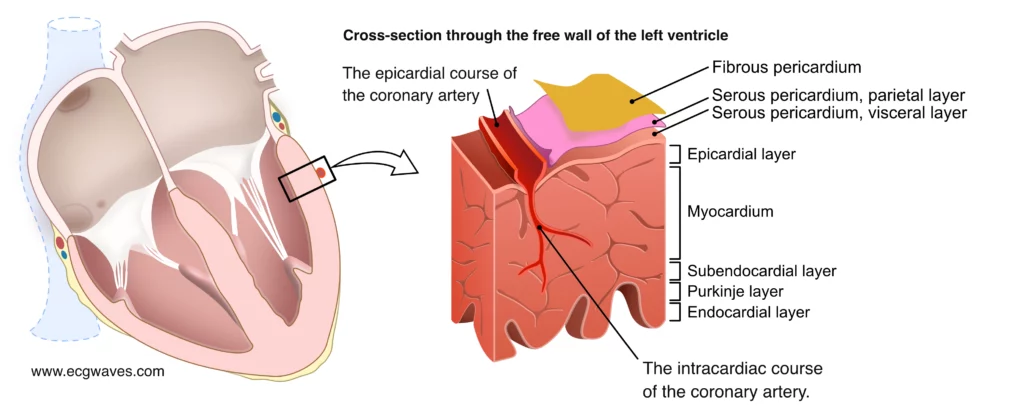
La figure 3 illustre la direction du flux sanguin et la progression de la nécrose. Le sous-endocarde est intrinsèquement plus vulnérable à l’ischémie pour plusieurs raisons anatomiques et physiologiques :
- Perfusion diastolique exclusive : Le myocarde ventriculaire gauche n’est perfusé que pendant la diastole. Les vaisseaux perforants traversent l’épaisseur du muscle de l’épicarde vers l’endocarde.
- Pression intramurale : Le sous-endocarde subit la plus forte pression compressive par la cavité ventriculaire, ce qui augmente la résistance vasculaire extravasculaire et limite la perfusion, surtout si la pression diastolique intra-ventriculaire (LVEDP) est élevée.
- Demande métabolique : Le stress pariétal (et donc la consommation d’oxygène) est plus élevé dans le sous-endocarde.
C’est pourquoi, en cas de sténose coronarienne limitant le flux, l’ischémie apparaît d’abord et préférentiellement dans cette couche profonde, même si l’artère épicardique reste perméable.
Phénomènes post-ischémiques : Sidération et Hibernation
Après un épisode ischémique, le myocarde peut présenter des dysfonctionnements contractiles durables même si les cellules sont vivantes (viables). La distinction est cruciale pour la décision de revascularisation :
- Le myocarde sidéré (« Stunned myocardium ») : Il s’agit d’une dysfonction contractile mécanique persistante après reperfusion, suite à un épisode ischémique aigu transitoire (ex: après thrombolyse réussie ou angioplastie primaire). Bien que le flux sanguin soit normalisé, les cellules restent « étourdies » pendant des jours ou des semaines avant de récupérer spontanément leur contractilité.
- Le myocarde hibernant : Il s’agit d’une adaptation à une ischémie chronique sévère (hypoperfusion chronique). Pour survivre avec un apport d’oxygène réduit, les myocytes réduisent leur contractilité au strict minimum. Contrairement à la sidération, l’hibernation ne récupère pas spontanément sans revascularisation (pontage ou stent) pour restaurer un flux adéquat. La détection de la viabilité (par IRM ou scintigraphie) est essentielle ici.