Le modèle en trois phases de l’arrêt cardiaque soudain
L’évolution physiopathologique de l’arrêt cardiaque soudain (ACS) vers le décès biologique n’est pas un événement instantané, mais un processus dynamique et progressif. Ce continuum affecte considérablement l’efficacité des thérapeutiques de réanimation. Une littérature abondante démontre que la réponse aux interventions (défibrillation, compressions, pharmacologie) est intrinsèquement sensible au temps écoulé depuis le collapsus (le « no-flow » et le « low-flow »). Pour maximiser la probabilité de rétablir une circulation spontanée (RACS ou ROSC), les stratégies devraient idéalement être adaptées à la phase physiopathologique en cours. Toutefois, les recommandations internationales actuelles (ILCOR, ERC, AHA) privilégient la simplicité opérationnelle et ne modulent pas systématiquement l’algorithme en fonction de la durée estimée de l’arrêt (hormis la priorité absolue à la défibrillation immédiate sur un rythme choquable observé). Cette approche standardisée s’explique par la difficulté pratique, en situation d’urgence, de déterminer avec précision la phase métabolique du patient. Néanmoins, la compréhension du modèle triphasique décrit par Weisfeldt et Becker (2002) est indispensable pour le clinicien, car elle sous-tend les mécanismes de l’échec de la réanimation et oriente les recherches actuelles vers des thérapies plus ciblées (tableau 1).
| Phase | Période (estimée) | Stratégie physiopathologique optimale |
|---|---|---|
| Phase électrique | 0 à 4 minutes | Défibrillation immédiate |
| Phase circulatoire | 4 à 10 minutes | « Priming » cardiaque : Compressions et ventilation, suivies d’une défibrillation |
| Phase métabolique | >10 minutes | Support extracorporel (E-CPR), contrôle de la reperfusion (hypothermie, etc.) |
La validité de ces phases est étayée par un large corpus d’études expérimentales animales et de données cliniques. La phase électrique et la phase circulatoire sont dominées par l’épuisement énergétique progressif et les altérations ioniques du myocyte. La phase métabolique, plus complexe, est caractérisée par une défaillance systémique multifactorielle incluant l’ischémie globale, l’acidose intracellulaire sévère, l’inflammation systémique (syndrome post-arrêt) et, paradoxalement, les lésions induites par la reperfusion.
Demande en oxygène du myocarde et bioénergétique
Le myocarde est un tissu métaboliquement exigeant qui fonctionne quasi exclusivement en aérobie. L’oxygène est indispensable à la phosphorylation oxydative mitochondriale pour la synthèse de l’adénosine triphosphate (ATP), le « carburant » cellulaire universel. Le cœur humain consomme et resynthétise environ 6 à 30 kilogrammes d’ATP par jour selon l’effort (Neubauer et al.), ce qui représente plusieurs multiples de son propre poids. Cette prouesse bioénergétique repose sur une densité mitochondriale exceptionnelle, ces organites occupant près de 30 % du volume cardiomyocytaire (Page et al.). Cette capacité permet un renouvellement complet du pool d’ATP intracellulaire toutes les 10 secondes environ (Fell et al., Houtkooper et al.). L’essentiel de cet ATP alimente l’interaction actine-myosine pour la contraction et, de manière cruciale, les pompes ioniques (Na+/K+ ATPase, SERCA) qui maintiennent les gradients électrochimiques vitaux.
En cas d’arrêt circulatoire, l’apport en oxygène s’effondre instantanément. Neumar et al. ont démontré la cinétique de l’effondrement énergétique : la concentration d’ATP chute drastiquement pendant la fibrillation ventriculaire (FV). Les biopsies myocardiques en série montrent qu’après seulement 5 minutes de FV non traitée, les réserves d’ATP sont amputées de 50 %, et la glycolyse anaérobie ne suffit pas à compenser la demande, entraînant une accumulation rapide de lactate et de protons (acidose).

L’épuisement de l’ATP a des conséquences catastrophiques immédiates : arrêt des pompes ioniques, surcharge calcique cytosolique et perte du potentiel de membrane. L’excitabilité électrique devient erratique et la fonction contractile s’arrête (sidération myocardique). Si la reperfusion n’intervient pas, la mort cellulaire par nécrose de coagulation débute typiquement dans les 20 minutes suivant l’arrêt circulatoire complet, marquant le passage vers des lésions irréversibles.
La phase électrique (0-4 min) : La priorité absolue à la défibrillation
La majorité des arrêts cardiaques extra-hospitaliers d’origine cardiaque débutent par une tachyarythmie ventriculaire, soit une tachycardie ventriculaire (TV) dégénérant en fibrillation ventriculaire (FV). Durant les premières minutes (phase électrique), le myocarde conserve des réserves d’ATP suffisantes et un environnement métabolique relativement préservé. La défibrillation électrique est alors le seul traitement efficace et doit être immédiate. L’efficacité des défibrillateurs automatiques implantables (DAI) illustre parfaitement ce concept : programmés pour choquer dans les 10 à 20 secondes, ils terminent l’arythmie dans près de 98 % des cas (Zipes et al., Volosin et al.).
En milieu clinique ou extra-hospitalier, le facteur temps est le déterminant majeur du pronostic. Le taux de survie avoisine les 70 % si le choc est délivré dans les 3 minutes (Hessulf et al.). Malheureusement, la réalité logistique des secours d’urgence impose souvent des délais médians d’arrivée de 10 à 15 minutes, période durant laquelle le patient glisse hors de la phase électrique. À ce stade tardif, la survie chute dramatiquement autour de 20-30 %, même si le rythme reste initialement choquable (Rawshani et al.).
| Délai entre l’apparition de la TV/FV et la défibrillation | Source des données | Taux de réussite de la cardioversion/défibrillation |
|---|---|---|
| <30 secondes | Essais cliniques DAI | 98 % |
| 3 minutes | Études d’observation (milieu hospitalier/témoins) | 70 % |
| >10-15 minutes | Études d’observation (secours extra-hospitaliers) | 20-30 % |
Chaque minute sans RCP ni défibrillation réduit la survie de 10 %. Le myocarde évolue d’un état de fibrillation « grossière » (haute amplitude, réserves énergétiques présentes) vers une fibrillation « fine », prélude à l’asystolie. La Figure 2 illustre cette fenêtre d’opportunité critique : au-delà de 4-5 minutes, le bénéfice d’un choc immédiat sans préparation hémodynamique préalable devient discutable.
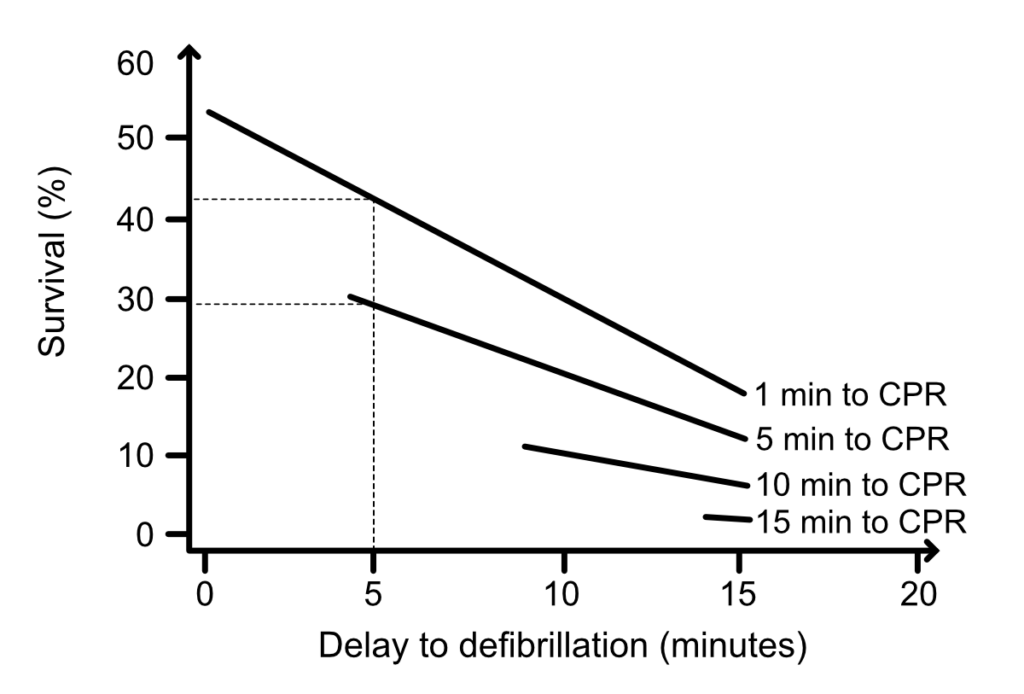
En résumé, durant la phase électrique (0-4 minutes), le myocarde est prêt à être défibrillé. Les compressions thoraciques, bien que jamais nuisibles, sont secondaires par rapport à l’urgence du choc électrique. C’est le fondement de l’utilisation des défibrillateurs automatisés externes (DAE) par le grand public.
La phase circulatoire (4-10 min) : Comprimer pour « recharger » le cœur
Lorsque l’arrêt se prolonge au-delà de 4 minutes, le cœur entre dans la phase circulatoire. L’épuisement en oxygène et en substrats énergétiques rend le myocarde ischémique rigide et réfractaire à la défibrillation. Un choc électrique délivré sur un cœur métaboliquement épuisé a de fortes chances d’induire une asystolie ou une activité électrique sans pouls (AESP) post-choc, plutôt qu’un rythme perfusant.
Dès 1943, Gurvich et al. suggéraient que la réanimation par compressions thoraciques avant le choc améliorait le succès de la défibrillation après une ischémie prolongée. Les compressions thoraciques ne servent pas uniquement à perfuser le cerveau ; elles sont cruciales pour générer une Pression de Perfusion Coronaire (PPC). La PPC permet un flux sanguin minime mais critique vers le myocarde, apportant l’oxygène nécessaire à la resynthèse partielle d’ATP et au « lavage » des métabolites toxiques. C’est ce qu’on appelle le « priming » cardiaque.
Les preuves expérimentales soutiennent cette approche séquentielle (Compressions d’abord, Choc ensuite) pour les arrêts prolongés :
- Yakaitis et al. ont montré qu’après 5 minutes de FV, le taux de succès de la défibrillation seule chute à 30 %, contre 70 % si elle est précédée de compressions.
- Niemann et al. rapportent qu’après 7 minutes de FV, l’efficacité du choc est triplée par une période préalable de RCP.
- Cobb et al. ont observé chez l’humain une amélioration de la survie de 42 % en imposant 90 secondes de RCP avant l’analyse du rythme lorsque le délai d’intervention dépassait 4 minutes.
Ces données soulignent que la qualité des compressions est primordiale. Toute interruption des compressions fait chuter la PPC à zéro quasi instantanément. Il faut ensuite plusieurs compressions consécutives pour rétablir une pression efficace. L’étude de Yu et al. est édifiante : une interruption pré-choc de plus de 15 secondes annule totalement les chances de réussite de la défibrillation dans un modèle animal ischémique. C’est la raison physiologique pour laquelle les directives actuelles insistent sur la réduction des temps de « main-levée » (hands-off time).
La phase métabolique (>10 min) et les lésions de reperfusion
Au-delà de 10 minutes d’arrêt sans RCP efficace, on entre dans la phase métabolique. Le pronostic devient sombre avec des taux de survie très faibles via les méthodes conventionnelles. À ce stade, l’ischémie sévère a entraîné une défaillance généralisée des barrières cellulaires, une translocation bactérienne intestinale potentielle et une activation massive des cascades inflammatoires (cytokines).
Le myocarde est en contracture ischémique (cœur de pierre) et en asystolie. Paradoxalement, la réintroduction brutale d’oxygène par la RCP à ce stade peut aggraver les dommages tissulaires : c’est le phénomène des lésions de reperfusion. Ce mécanisme implique :
- Le stress oxydatif : La production massive d’espèces réactives de l’oxygène (ROS) lors de la réoxygénation attaque les membranes cellulaires (peroxydation lipidique).
- La surcharge calcique : L’incapacité du réticulum sarcoplasmique à recapturer le calcium entraîne une hypercontracture et l’ouverture du pore de transition de perméabilité mitochondriale (mPTP), déclenchant l’apoptose.
- Le phénomène de « no-reflow » : L’œdème endothélial et l’agrégation plaquettaire empêchent la perfusion microvasculaire même si la circulation épicardique est rétablie.
«
Pendant cette phase critique, les stratégies standards (ACLS/ALS) atteignent leurs limites. L’administration répétée d’adrénaline peut être délétère en augmentant la consommation d’oxygène myocardique et en aggravant les troubles de la microcirculation cérébrale. C’est pourquoi la recherche se tourne vers des approches plus invasives pour ces patients.
Implications thérapeutiques modernes : ECPR et protection d’organes
Contrairement à l’idée que le traitement est « inconnu » pour la phase métabolique, la médecine cardiovasculaire moderne a développé des stratégies de sauvetage pour ces arrêts réfractaires. L’assistance circulatoire extracorporelle (E-CPR ou VA-ECMO) permet de restaurer une perfusion et une oxygénation systémiques indépendantes du cœur natif défaillant. Cela offre une fenêtre de temps pour traiter la cause réversible (souvent une occlusion coronaire nécessitant une angioplastie) tout en mettant le cœur au repos.
De plus, la prise en charge post-RACS (Post-Cardiac Arrest Syndrome) est focalisée sur l’atténuation des lésions de reperfusion, notamment par le contrôle ciblé de la température (Targeted Temperature Management – TTM) pour neuroprotéger le cerveau, et le maintien d’une hémodynamique stable pour éviter l’hypoperfusion secondaire. La compréhension du modèle triphasique ne change pas seulement l’approche per-arrêt, mais justifie l’agressivité des soins post-réanimation.
Références
Weisfeldt ML, Becker LB. Resuscitation after cardiac arrest: a 3-phase time-sensitive model. JAMA. 2002;288:3035-8.
Kaustubha D. Patil, Henry R. Halperin, Lance B. Becker. Cardiac Arrest: Resuscitation and Reperfusion. Circulation Research. 2015;116:2041–2049.
Neubauer S. The failing heart-An engine out of fuel. New England Journal of Medicine. 2007;356:1140-1151.
Fell DA, Sauro HM. Metabolic control analysis. The effects of high enzyme concentrations. European Journal of Biochemistry. 1990;192:183-187.
Houtkooper RH, Canto C, Wanders RJ, Auwerx J. The secret life of NAD: An old metabolite controlling new metabolic signaling pathways. Endocrine Reviews. 2010;31:194-223.
Gurvich NL, Yuniev GS. Restoration of Heart Rhythm during Fibrillation by a Condenser Discharge. Am Rev Sov Med. 1947;4:252-6.
Vanden Hoek TL, Shao Z, Li C, Zak R, Schumacker PT, Becker LB. Reperfusion injury in cardiac myocytes after simulated ischemia. Am J Physiol. 1996;270:H1334-H1341.
Yakaitis RW, Ewy GA, Otto CW, Taren DL, Moon TE. Influence of time and therapy on ventricular defibrillation in dogs. Crit Care Med. 1980;8:157-163.
Menegazzi JJ, Davis EA, Yealy DM, et al. An experimental algorithm versus standard advanced cardiac life support in a swine model of out-of-hospital cardiac arrest. Ann Emerg Med. 1993;22:235-239.
Cobb LA, Fahrenbruch CE, Walsh TR, et al. Influence of cardiopulmonary resuscitation prior to defibrillation in patients with out-of-hospital ventricular fibrillation. JAMA. 1999;281:1182-1188.
Niemann JT, Cairns CB, Sharma J, Lewis RJ. Treatment of prolonged ventricular fibrillation. Circulation. 1992;85:281-287.
Rawshani A, et al. survival in Out-of-Hospital Cardiac Arrest. N Engl J Med. 2015.
Yu T, Weil MH, Tang W, et al. Adverse outcomes of interrupted precordial compression during automated defibrillation. Circulation. 2002;106:368-372.
Garcia LA, Allan JJ, Kerber RE. Interactions between CPR and defibrillation waveforms. Resuscitation. 2000;47:301-305.
Page E, McCallister LP. Quantitative electron microscopic description of heart muscle cells. Application to normal, hypertrophied and thyroxin-stimulated hearts. Am J Cardiol. 1973;31:172-181.