Un nouveau syndrome d’arythmie cardiaque potentiellement mortel identifié grâce à l’ECG
L’ECG peut être utilisé pour diagnostiquer plusieurs maladies cardiaques génétiques, telles que le syndrome de Brugada, le syndrome du QT long [LQTS] et le syndrome de repolarisation précoce. Ces affections ont une base génétique sous-jacente et peuvent provoquer des arythmies mortelles. Le syndrome du QT long a été identifié il y a plusieurs décennies par Jervell et al. Le syndrome de Brugada a été identifié au début des années 1990 et la repolarisation précoce a été identifiée comme un facteur de risque d’arrêt cardiaque soudain en 2008 (1, 2, 3). En novembre 2018, des chercheurs du Danemark, des Pays-Bas et du Royaume-Uni ont signalé un nouveau syndrome ECG caractérisé par des dépressions généralisées du segment ST et un risque accru d’arrêt cardiaque soudain. Ils ont identifié cinq familles non apparentées présentant des caractéristiques qui représentent un syndrome autosomique dominant non reconnu auparavant (4).
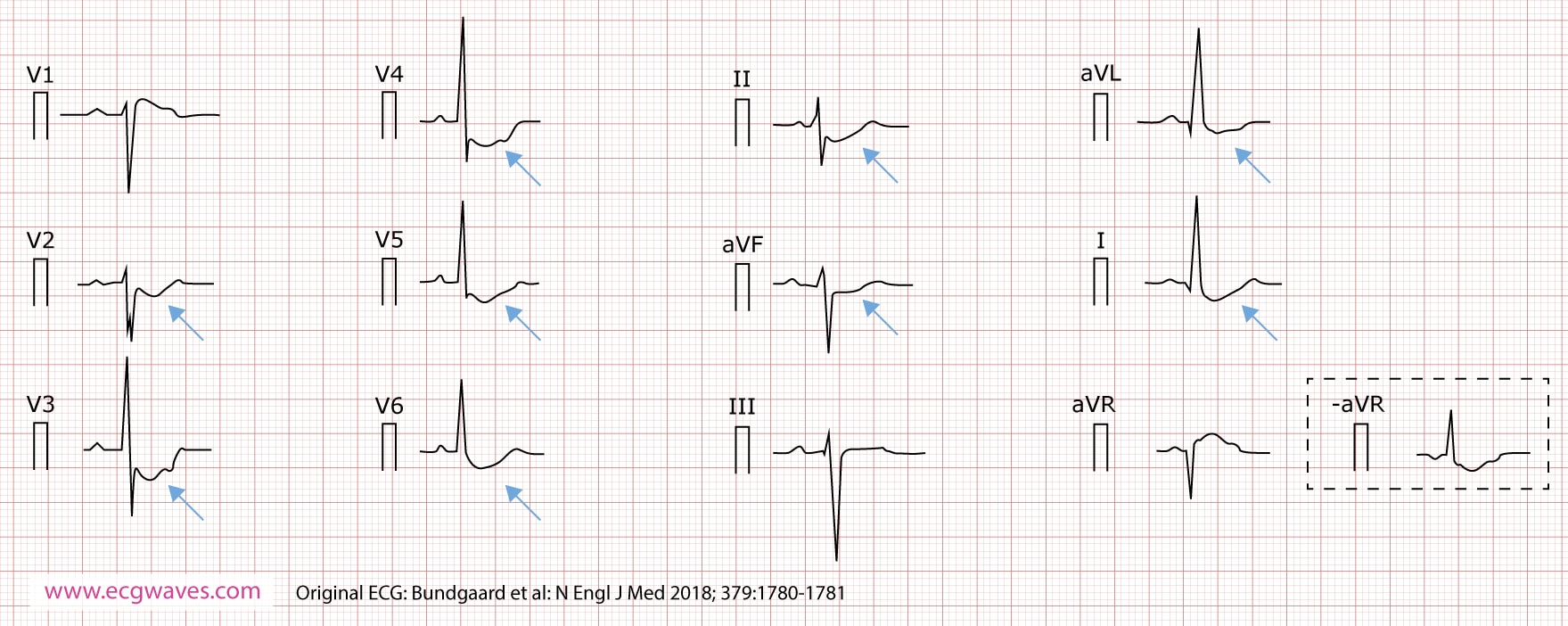
Un patient de sexe masculin (figure 1) s’est présenté à l’âge de 36 ans avec un ECG montrant une dépression profonde et persistante du segment ST, concave vers le haut, dans les dérivations I, II, aVL, aVF, et V2 à V6. Ces modifications de l’ECG sont restées stables pendant 30 ans. Les dépressions du segment ST étaient accentuées pendant l’exercice. Le patient a développé une fibrillation auriculaire à l’âge de 55 ans. À l’âge de 64 ans, il a développé une fibrillation ventriculaire et a avorté d’une mort cardiaque subite. Son fils et sa fille ont présenté des modifications similaires de l’ECG. Quatre autres membres de la famille ont présenté des modifications similaires de l’ECG. Deux membres de la famille sont décédés d’une mort cardiaque subite. Une fibrillation auriculaire et des arythmies ventriculaires ont été observées dans la cinquième décennie de la vie chez plusieurs membres de la famille. Les examens d’imagerie n’ont pas révélé d’anomalies cardiaques structurelles, ni de maladie coronarienne.
Un autre patient de sexe masculin, présentant des modifications similaires du segment ST, a connu des épisodes syncopaux, des tachycardies ventriculaires (y compris des torsades de pointes), des arrêts de fibrillation ventriculaire et un dysfonctionnement ventriculaire gauche modéré. Ce patient est décédé à l’âge de 17 ans des suites de complications liées à son arythmie. Son père et son oncle, qui présentaient des modifications similaires du segment ST, ont été diagnostiqués avec une fibrillation auriculaire à l’âge de 60 ans. Le père a connu des épisodes répétés de tachycardie ventriculaire et de fibrillation ventriculaire. Plusieurs membres de la famille ont présenté des modifications identiques de l’ECG (le plus jeune à l’âge de 6 ans).
Neuf autres personnes atteintes ont été présentées dans le cadre de l’étude. Tous les tests génétiques, utilisant des panels de gènes pour les mutations connues des maladies cardiovasculaires, se sont révélés négatifs.
Résumé de l’étude
- Bundgaard et al ont vraisemblablement identifié un nouveau syndrome cardiaque familial (autosomique dominant). L’anomalie génétique sous-jacente reste à découvrir.
- Le diagnostic de ce syndrome repose sur l’ECG, les antécédents familiaux et la présentation clinique.
- Les patients présentent une syncope, une fibrillation auriculaire précoce, des arythmies ventriculaires et un arrêt cardiaque soudain.
- Compte tenu de la prévalence des arythmies auriculaires, des arythmies ventriculaires et des arrêts cardiaques soudains, ce syndrome semble très grave.
- L’ECG se caractérise par une dépression du segment ST marquée, persistante et non ischémique. Ces modifications du segment ST sont stables dans le temps (contrairement au syndrome de Brugada et au STLQ, qui présentent tous deux des modifications très dynamiques de l’ECG).
Sujets connexes
Modifications du segment ST et de l’onde T
Dépression ischémique et non ischémique du segment ST
Auteur de l’article
Dr Araz Rawshani, MD, PhD
Université de Göteborg, Hôpital universitaire Sahlgrenska, Suède
Références
- Jervell A, Lange-Nielsen F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death (Surdité congénitale, cardiopathie fonctionnelle avec allongement de l’intervalle Q-T et mort subite). Am Heart J 1957;54:59-68.
- Brugada P, Brugada J. Bloc de branche droit, élévation persistante du segment ST et mort cardiaque subite : un syndrome clinique et électrocardiographique distinct – un rapport multicentrique. J Am Coll Cardiol 1992;20:1391-1396.
- Haïssaguerre M, Derval N, Sacher F, et al. Sudden cardiac arrest associated with early repolarization. N Engl J Med 2008;358:2016-2023.
- Bundgaard et al. Un nouveau syndrome familial d’arythmie cardiaque avec dépression généralisée du segment ST. N Engl J Med 2018 ; 379:1780-1781.